|
Dna测序释疑
1、DNA测序样品用什么溶液溶解比较好?
答:溶解DNA测序样品时,用灭菌蒸馏水溶解最好。DNA的测序反应也是Taq酶的聚合反应,需要一个最佳的酶反应条件,如果DNA用缓冲液溶解后,在进行了测序反应时,DNA溶液中的缓冲液组份会影响测序反应的体系条件,造成Taq酶的聚合性能下降。 有很多客户在溶解DNA测序样品时使用TE Buffer。虽然TE Buffer能增加DNA样品保存期间的稳定性, 但TE Buffer对DNA测序反应有影响,根据我们的经验,我们还是推荐使用灭菌蒸馏水来溶解DNA测序样品。
2、提供DNA测序样品时,提供何种形态的比较好?
答:我们推荐客户提供菌体,由我们来提取质粒,这样DNA样品比较稳定。 如果您要以提供DNA样品,我们也很欢迎,但一定要注意样品纯度和数量。 提供的测序样品为PCR产物时,特别需要注意DNA的纯度和数量。PCR产物应该进行切胶回收,否则无法得到良好的测序效果。 有关DNA测序样品的详细情况请严格参照“测序模板的要求”部分的说明。
3、提供的测序样品为菌体时,以什么形态提供为好?
答:一般菌体的形态有:平板培养菌、穿刺培养菌,甘油保存菌或新鲜菌液等。我们提倡寄送穿刺培养菌或新鲜菌液。 平板培养菌运送特别不方便,我们收到的一些平板培养菌的培养皿在运送过程中常常已经破碎,面目全非,需要用户重新寄样。这样既误时间,又浪费客户的样品。一 旦是客户非常重要的样品时,其后果更不可设想。而甘油保存菌则容易污染。 制作穿刺菌时,可在1.5ml的Tube管中加入琼脂培养基,把菌体用牙签穿刺于琼脂培养基(固体)中,37℃ 培养一个晚上后便可使用。穿刺培养菌在4℃下可保存数个月,并且不容易污染,便于运送。再一夏天运输样品时一定要用冰盒冰袋。
4、与测序引物有关的问题:
答:对于通用测序引物,只要正确使用,一般不会有太大问题,测序引物问题主要发生在客户自己提供的PCR引物上。应该明确的一点是并不是所用的用于PCR的引物都可以用来作测序,以下几种PCR引物将是不适 合用作测序引物的:
◆简并引物, 简并引物必然要在测序模板上有多个结合位点,直接影响测序结果。
◆随机引物,如RAPD引物, 随机引物一般都比较短,所用退火温度低,在测序反应的条件下,不能很好地与模板结合。
◆过长的引物,一般要求测序引物不大于24bp,最长不能超过30bp。 过长的引物在测序反应的较低的条件下容易在测序模板上有多个结合位点,导致测序结果背景增高。另外,较长的引物纯度也将难以保证。通常用于测序的引物纯度要在90%以上,引物纯度低时,测序反应的背景将明显增大,直接影响到测序结果。
◆有特殊标记的引物,该情况主要指荧光标记的引物。我们测序反应的四种碱基都是荧光标记的,这样,荧光标记的引物将产生干扰。另外,其他一些有大的标记基团的引物也最好不要用于测序。引物上大的标记基团将直接影响到DNA 片段的迁移率,导致测序结果峰型不好或错误。
◆不纯的引物, 测序引物对纯度的要求很高,合成的引物中非全长的片段可以造成较强的背景。以一个20bp的测序引物为例, 直接脱盐纯化的话,纯度至多在70%左右,也就是说将有30%的引物将作为背景噪音,这必将严重影响测序结果。 一般经PAGE或OPC法纯化的引物基本能达到测序的要求。
5、怎样选择(设计)测序用引物?
答:测序用引物要求非常严格,不同于PCR用引物。PCR用引物一般只要能和模板结合,3’端的几个碱基能完 全配对,即使引物长达80~100多个碱基,只要调整PCR反应条件,也能成功进行PCR反应。 而测序用引物便不一样了,必须严格符合以下要求。本公司的测序用引物全用引物设计软件Primer设计。在本公司测序时,我们可免费帮助设计测序用引物。
◆长度在15~25个碱基左右,一般选择20个碱基(根据GC含量作适当调整)。
◆3’端尽量选择G或C碱基(但不绝对),以增加与模板的结合能力。
◆Tm温度应选择50℃~70℃左右。
◆GC含量应选择在50%左右,尽量避开A、T、G、C的连续结构。
◆避开引物自身形成发夹结构或引物二聚体结构等复杂结构。
◆保证引物和模板100%匹配,特别是3’端的几个碱基一定要100%匹配。同时必须严格保证引物和模板之间只能有一个结合位点。
6、PCR片段直接测序和PCR片段经克隆后测序的结果有何区别?
答:众所周知,PCR圹增过程中会出现很多错配现象,但不可能所有的错配都发生在同一位置。PCR片段直接测序时,其结果是PCR片段众多分子的混合物的结果。如果在某一个点上出现了几十次错配现象,但大多数 分子(或许是几十万个分子)在这个点上应该还是正确的,在测序时,错配现象也就是反映不出来了。因此, PCR片段直接测序的结果反映的是PCR用模板最原始的结果。而PCR片段经克隆后测序是测定了某一个分子的DNA序列。在几十个循环的PCR扩增过程中,很难保证某一个分子的任何点都不发生错配。因此,PCR片段经克隆后的测序结果,往往存在着一些错配的序列,和PCR片段直接测序的结果相比有些碱基会有所不同。这种错配现象的多少取决于PCR扩增时使用的DNA聚合酶的保真性能。要减少PCR扩增过程中的错配现象,在PCR反应时,请选用保真性能高的DNA聚合酶。
7、测序结果不到800BP还照常收费了,为什么?
答:如在DNA样品中的DNA序列分布匀称,没有复杂结构时,正常的测序反应能保证达到800Bases以上。但有 一些DNA样品立体结构复杂,造成聚合酶延伸反应终止,测序信号突然减弱或消失,或者测序结果出现套峰现象,出现这些现象的原因由DNA模板本身所造成,如果是由于本中心保证进行调整测序。对这种情况,本中心会根据具体情况
收费(详细见收费约定)。出现这些情况的原因分析如下:
◆G/C rich、G/C Cluster。这种情况一般表现为测序信号突然减弱或消失(图1,图2)。
图1 GC引起的信号减弱
图2 GC引起的信号消失
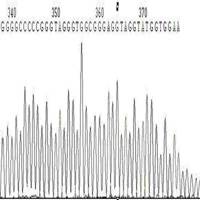
◆A,T的连续结构。这种情况一般表现为A、T连续结构后面的测序结果出现套峰。根据文献记载。原因 在于聚合酶进行聚合反应时,由于A或T的连续,聚合酶难以识别完整的每个A或T,在某个A或T的后面便开始进行A或T连续结构以后序列的聚合反应(打滑现象),造成测序结果紊乱,出现套峰。
一般在多少个A或T的后面能出现这种情况呢?现在还没有这方面的报道。根据我们的经验,这一情况的出现和A或T的连续结构后面的序列的排列情况有着直接的关系。有时10多个A或T的连续结构后面便出现套峰,但我中心测序有时60~70个A或T的连续结构后面的序列也一样可以完整地读出来。具体情况还有待考证。 一般来说,PCR片段直接测序时,A或T的连续结构后面的序列测序结果都会出现套峰。原因在于测序时经历了 PCR反应及测序反应(测序反应本身也是PCR反应)二次聚合酶的打滑现象。
◆ poly结构引起的套峰。poly结构有时测的很长有时却很短,三博最长达到120BP都没有问题。
图3 polyA引起的套峰
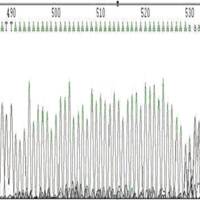
◆原因不明的复杂结构:测序结果出现突然信号减弱或消失。从序列上看,DNA碱基排列并无特别异常。 估计是DNA整体出现复杂结构,从某一位置开始聚合酶的聚合反应便无法进行。
图4 复杂结构引起的信号中断

8、出现套峰的原因是什么?
答:在测序反应中,模板或引物的原因都可能造成套峰的形成,归结其形成原因有以下几点:
◆测序引物在模板上有两个结合位点(图5);
◆模板不纯,如果是质粒或是菌液,原因是非单克隆(图6),如果是PCR原因为非特异性条带(图7);
◆模板序列的特殊结构,如poly结构、发卡结构等(图8);
◆引物降解,或引物不纯(图9,图10)。
图5 双引物结合位点引起的套峰
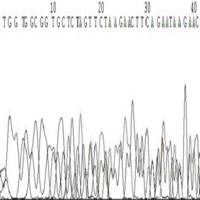
图6 由于质粒或菌液为非单克隆引起的套峰
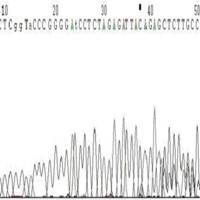
图7 PCR为非特异性条带引起的套峰

图8 模板特殊结构引起的套峰
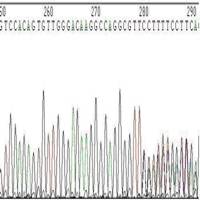
图9 引物轻微降解或引物不纯引起的套峰
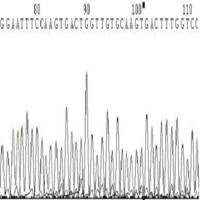
图10 引物严重降解或引物不纯引起的套峰
9、为什么找不到我的PCR引物?
答:以下几种情况,我们将无法找到做PCR时的引物序列 :
◆用PCR引物作为测序引物进行测序时,所测序列是从引物3’末端后第一个碱基开始的,所以自然就找不到您的引物序列了。有两种方法可以得到您的引物序列。对于较短的PCR产物(<800bp),可以用另一端的引物进行测序,从另一端测序可以一直测到序列的末端,就可以在序列的末端得到 您的引物的反向互补序列。对于较长的序列,一个测序反应测不到头,因此就只能将您的PCR产物片段克隆到适当的载体中,用载体上的通用引物进行测序。由于载体上的通用引物与您的插入序列之间还有一段距离,因此就可以得到您的完整的引物序列。由于在测序的起始端总会有一些碱基无法准确读出,因此,您如果想得到您的PCR产物的完整序列,最好克隆后进行测序。
◆PCR产物用T载体克隆后,由于克隆的方向是随机的,因此,当您在一条链上找不到您的引物序列时,试图在互补链上寻找您的引物序列。
◆当测序引物离您的插入片段很近时,有时可能也无法找到您的引物的全序列。这主要是因为有时测序的起始端由于未去除的染料或引物二聚体的干扰,造成起始区的序列不好,可能无法找到您的引物完整序列。
◆有时,质粒做模板进行测序时,由于某些原因,质粒上没有插入外援片段,为空载体,所测的序列完全为载体序列,此时自然也找不到引物序列。
10、我的基因序列与标准序列为什么有差别?
答:一段基因序列经扩增后,克隆到载体中进行测序。在两个层次上可能导致序列发生变化。首先在PCR扩增过程中就可能产生错误。将片段克隆到载体中也有可能发生突变。其次,测序的准确率问题。 ABI公司承诺其仪器的测序精度在一定范围内可以达到98.5%以上。由于仪器准确率的限制,在一个较 长的序列中发生碱基序列错误是难以避免的。在确认克隆无误的情况下,通过双向测序可以最大限度 减少测序的错误。您如果想得到您的最准确的序列,进行双向测序是很有必要的。只进行简单的单向 测序,我们无法保证所测序列的完全准确性,这是由仪器的精度决定的。
11、过短的PCR产物为什么不适于直接测序?
答:首先过短的PCR产物纯化困难,一般的PCR产物纯化试剂盒都要求PCR产物片段大于150bp,过短的 PCR产物纯化和准确定量都非常困难。因此我们要求用于测序的PCR产物一般不低于150bp长度。其次,由于测序技术本身的限制,测序反应对环境的干扰比较敏感,模板太短的PCR测序受外界的 干扰更大,很容易造成测序失败。
12、用测序的方法检测点突变可靠吗?
答:有的客户想用测序的方法检测点突变体,我们认为该方法可靠性不高。主要有以下两个原因。首先,我们并不清楚突变的序列与正常的序列的比例是多少。测序反应的信号强度直接与模板的量有关,如果突变的模板所占的比例很少,将直接作为背景噪音了,很难检测出来。只有当测序反应体系中正常的和突变的模板量比较接近时,才能较可靠地检测到突变体的存在。其次,在同一位置,不同碱基的信号强度一般是不一样的。这样即使突变的模板所占的比较较高时,也不一定能准确检测到突变的存在。另外,测序仪是设计用来测序正常的碱基序列的,软件在对扫描的结果进行处理时,会尽量提高主峰而将背景信号尽量压低,以得到尽可能好的结果。因此,当某处出现双峰时,测序仪一般会认为信号弱的峰为背景信号,在处理过程中,将弱的峰进一步压低,这样根部不立于突变体的检测。因此认为,用测序的方法检测突变体的存在不是一个好的方法。
13、我要求5’到3’正向测序,为什么你们给我的序列是反的?
答:您指的可能是插入片段的方向,而我们并不清楚您的样品是如何构建的。我们只能根据质粒上的 序列来确定测序方向,所以在测序引物一栏中请不要使用3’引物和5’引物这样的字样,因为我们手中的资料在注明方向时可能和您手中的资料方向相反,请以T7,T3,SP6,M13f,M13r这样的形式来 填写,或注明酶切位点方向比如“测序方向EcoRI到HindIII”。我们手中等质粒资料有限,有时还需要您提供质粒的相关资料。
14、我的样品在你们所说的可靠范围内有一处存在疑问,能否重新测一次?
答:我们希望您能够告诉我们存在疑问的位点的位置,如果从测序报告上的确无法作出准确判读,我们会重新进行实验。如果您指出的位点信号清晰准确,您仍然要求再进行一次实验,那么实验结果和验证实验是相同的。
15、我的样品送测序前已经鉴定过了,有插入片段的,为什么你给我的测序结果是一个空质粒?
答:造成这种现象主要有两个原因。
◆鉴定过程中的假阳性。鉴定插入片段主要是通过PCR和酶切两种方式鉴定。由于PCR反应受多种条件的影响,在鉴定过程中易产生假阳性,而酶切鉴定是比较可靠的鉴定方式。
◆我们由于条件的限制,无法单独的对某一个客户的样品进行特定条件的培养,所以可能在培养过程中发生插入片段的丢失。由于这种情况的发生无法事先预期到,所以我们也只能对出现这样情况的客户说抱歉。直接提供质粒虽然可以避免这样的情况,但由于质粒制备上的问题,测序的成功率可能会受到影响。
16、DNA片段很长,一个反应读不到头,怎么办?
答:如果DNA片段在载体上,可以用载体上两端的引物同时测序,让其中间交叉互补,便可完全测通。如果这样还读不通,可在已经测出序列的450~500个碱基处设计测序引物作进一步测序(此称为Primer Walking法),便可完全测序。
17、我的样品你们已经测通了,但为什么在overlap区有这么多的错配,给出的全序列和单个报告也称作差异,我该相信谁?
答:给出的全序列是一个拼接的结果,当互相拼接的两个序列存在差异时,应该以序列质量更好的应该为主,这也就是为什么会出现错配和全序列与单个测序结果的差异。
18、你们为什么在primer walking时总将引物设计的那么靠前?
答:我们在设计引物时有两个准则。一是设计引物区域的序列必须准确,二是在引物区后必须还有足 够的准确序列以便拼接。这样我们设计引物的位置就必然比较靠前,在满足软件设定的条件下,我们 总会选取最靠近3’端的引物来作为最终的测序引物。
19、我有一个4KB的PCR片段,希望你们帮我测通。
答:大于2KB的PCR片段要测通最好是构建好克隆后再进行测序。这样模板更加稳定,测序效果也更加好一些。
20、测序完成后,测序样品和引物将如何处理(或保存)?
答:客户需要将测序样品或引物(客户自带的)返还时,我们在发送测序报告的同时,按客户要求寄回样品或引物(客户自带的)。 对于没返回的测序样品和引物,公司负责保存1个月(从样品收到之日算起),超过1个月还需测序的样品,请客户另行提供。
21、造成测序结果中N值较多原因:
◆PCR产物直接进行测序,序列结束后无反应信号。PCR产物测序一般末端的一个碱基为A(绿色峰),这是由于Taq酶能够在PCR反应的末端非特异性地加上一个A碱基,我们用T载体克隆PCR产物就是根据该原理的,这样我们很容易辨别PCR产物结束的位点。在此序列以外就不是我们所要的序列了。
◆在序列的起始端有时会有一些N值。该种情况主要是有未去除的染料单体、样品小片段、引物二聚体的干扰造成。干扰峰和正常序列峰重叠在一起,仪器无法正确判断出为何碱基。本中心染料干扰问题基本解决。本中心测序一般20BP以后就可精确读取。
◆在序列的3’端容易产生N值。一个测序反应一般可以读出800-1000bp的碱基,如果样品好可以精确读取800bp,占总反应80%左右,但是,有20%只能保证600bp以前的碱基是可靠的,会有许多碱基分不开,就会产生N值,这种情况下产生的N值是无法避免的。如果有希望测长我们会调整重测。
◆测序模板本身含有杂合序列,该种情况主要发生在PCR产物直接测序上。由于PCR产物本身是一个混和溶液,相近的片断又很难割胶纯化出来,这样和引物结合就很容易产生系列杂合序列就会造成在某些位置上有套峰,整个测序信号都非常好,这是由于套峰产生的N值。该种情况明显是客户的样品问题。通过将PCR产物克隆后进行测序,可以解决该种情况的N值问题。
◆造成N值,大部分情况是客户样品的问题,如果是测序的问题我们保证免费重新调整测序一次。 | 